Code
library(ggplot2)
library(dplyr)
library(reshape2)
library(readr)
source("theme_donq.R")
timepoints <- c("Pre", "Wk3", "Wk6", "Wk9")Parse, visualize, and interpret NMF factors from MEGPath runs
Scripts 01–03 screened preprocessing approaches using R’s NMF package. The best candidates were then run through MEGPath — the production C++ NMF algorithm (Dyer & Dymacek 2018) that uses Monte Carlo Markov Chain + simulated annealing. MEGPath normalizes each input matrix to [0, 1] internally and minimizes LAD (Least Absolute Deviation = \(\sum|V - WH|\)).
We compiled MEGPath locally and ran 8 configurations at 100K iterations each:
| Configuration | Matrix | k | Rows |
|---|---|---|---|
| scRNA Baseline | Ethan’s dense_dataLT.csv (double-log, all 17,857 genes) |
2, 3 | 17,857 |
| scRNA Improved | SCTransform + 2,000 HVGs | 2, 3 | 2,000 |
| TCR Baseline | Ethan’s tcr_expansion.csv (all 2,541 clonotypes) |
2, 3 | 2,541 |
| TCR Improved | Bio Filter C (Expanded \(\cup\) TIL-observed, 416 clonotypes) | 2, 3 | 416 |
MEGPath is installed permanently at megpath/ within this project (with Eigen 3.4.0 bundled at megpath/eigen-3.4.0/). To rerun or add new configurations:
# MinGW must be in PATH for runtime libraries
export PATH="/c/Users/mattj/AppData/Local/Microsoft/WinGet/Packages/BrechtSanders.WinLibs.POSIX.UCRT_Microsoft.Winget.Source_8wekyb3d8bbwe/mingw64/bin:$PATH"
# Run from megpath_runs/ directory
cd megpath_runs
../megpath/standard/connmf.exe -a args_tcr_baseline_k2.txtArgument files specify the input CSV, rank (via one_patterns — number of empty strings = k), output directory, and iteration count. Pre-create the output directory before running (mkdir -p ./output_dir/). Set stats = "all" or no output files are written. File paths must not contain spaces (MEGPath crashes on them).
To recompile after modifying source:
cd megpath/shared && mingw32-make clean && mingw32-make
cd ../standard && mingw32-make clean && mingw32-makeThis script parses the MEGPath output files, extracts the H matrix (temporal patterns: how each factor behaves across the 4 timepoints) and W matrix (gene/clonotype loadings: which features belong to each factor), and interprets the results biologically.
library(ggplot2)
library(dplyr)
library(reshape2)
library(readr)
source("theme_donq.R")
timepoints <- c("Pre", "Wk3", "Wk6", "Wk9")MEGPath output format:
#): metadata + H matrix (patterns \(\times\) timepoints), space-separatedrow-id, W[0], W[1], ..., W[k-1], row_error — W matrix coefficients followed by the per-row reconstruction errorparse_megpath <- function(filepath) {
lines <- readLines(filepath)
# --- Metadata ---
meta <- list()
meta$file <- gsub("^#File : ", "", lines[grep("^#File", lines)])
meta$n_genes <- as.integer(gsub("^#Number_of_genes : ", "", lines[grep("^#Number_of_genes", lines)]))
meta$max_runs <- as.integer(gsub("^#MAX_RUNS : ", "", lines[grep("^#MAX_RUNS", lines)]))
meta$k <- as.integer(gsub("^#Num_Patterns : ", "", lines[grep("^#Num_Patterns", lines)]))
meta$total_error <- as.numeric(gsub("^#Total_error : ", "", lines[grep("^#Total_error", lines)]))
meta$runtime <- gsub("^#Total_running_time : ", "", lines[grep("^#Total_running_time", lines)])
# --- H matrix (patterns) ---
# Lines like: #"",0.464416 0.464727 0.46484 0.464863
h_lines <- grep('^#"",', lines, value = TRUE)
H <- do.call(rbind, lapply(h_lines, function(l) {
vals <- gsub('^#"",', '', l)
as.numeric(strsplit(trimws(vals), "\\s+")[[1]])
}))
colnames(H) <- c("Pre", "Wk3", "Wk6", "Wk9")
rownames(H) <- paste0("Factor_", 1:nrow(H))
# --- W matrix (coefficients) + per-row error ---
data_lines <- grep("^row-", lines, value = TRUE)
parsed <- do.call(rbind, lapply(data_lines, function(l) {
parts <- strsplit(l, ",")[[1]]
as.numeric(parts[-1]) # drop row-id
}))
k <- meta$k
W <- parsed[, 1:k, drop = FALSE]
row_errors <- parsed[, k + 1]
colnames(W) <- paste0("Factor_", 1:k)
list(meta = meta, H = H, W = W, row_errors = row_errors)
}runs <- list(
scrna_baseline_k2 = "megpath_runs/scrna_baseline_k2/one_results.csv",
scrna_baseline_k3 = "megpath_runs/scrna_baseline_k3/one_results.csv",
scrna_sct2k_k2 = "megpath_runs/scrna_sct2k_k2/one_results.csv",
scrna_sct2k_k3 = "megpath_runs/scrna_sct2k_k3/one_results.csv",
tcr_baseline_k2 = "megpath_runs/tcr_baseline_k2/one_results.csv",
tcr_baseline_k3 = "megpath_runs/tcr_baseline_k3/one_results.csv",
tcr_bio_k2 = "megpath_runs/tcr_bio_k2/one_results.csv",
tcr_bio_k3 = "megpath_runs/tcr_bio_k3_full/one_results.csv"
)
results <- lapply(runs, parse_megpath)summary_df <- do.call(rbind, lapply(names(results), function(name) {
m <- results[[name]]$meta
data.frame(
Run = name,
Data = ifelse(grepl("scrna", name), "scRNA", "TCR"),
Matrix = ifelse(grepl("baseline", name), "Baseline", "Improved"),
k = m$k,
Rows = m$n_genes,
LAD = round(m$total_error, 1),
LAD_per_element = round(m$total_error / (m$n_genes * 4), 5),
Runtime = m$runtime,
stringsAsFactors = FALSE
)
}))
knitr::kable(summary_df, caption = "MEGPath Production Results (100K iterations)")| Run | Data | Matrix | k | Rows | LAD | LAD_per_element | Runtime |
|---|---|---|---|---|---|---|---|
| scrna_baseline_k2 | scRNA | Baseline | 2 | 17857 | 452.0 | 0.00633 | 0d0h26m35.285s |
| scrna_baseline_k3 | scRNA | Baseline | 3 | 17857 | 237.3 | 0.00332 | 0d0h42m28.254s |
| scrna_sct2k_k2 | scRNA | Improved | 2 | 2000 | 14.1 | 0.00176 | 0d0h2m51.015s |
| scrna_sct2k_k3 | scRNA | Improved | 3 | 2000 | 14.1 | 0.00177 | 0d0h4m25.178s |
| tcr_baseline_k2 | TCR | Baseline | 2 | 2541 | 20.5 | 0.00202 | 0d0h3m38.207s |
| tcr_baseline_k3 | TCR | Baseline | 3 | 2541 | 20.2 | 0.00199 | 0d0h5m44.855s |
| tcr_bio_k2 | TCR | Improved | 2 | 416 | 5.4 | 0.00327 | 0d0h0m35.94s |
| tcr_bio_k3 | TCR | Improved | 3 | 416 | 5.2 | 0.00315 | 0d0h0m55.798s |
plot_df <- summary_df %>%
mutate(Label = paste0(Matrix, " k=", k),
Label = factor(Label, levels = unique(Label)))
ggplot(plot_df, aes(x = Label, y = LAD_per_element, fill = Matrix)) +
geom_col(width = 0.6) +
geom_text(aes(label = sprintf("%.5f", LAD_per_element)),
vjust = -0.5, color = "#b0b0b0", size = 3.5) +
facet_wrap(~Data, scales = "free") +
scale_fill_manual(values = c("Baseline" = "#555555", "Improved" = "#ffffff")) +
labs(title = "MEGPath Error per Element: Baseline vs Improved",
subtitle = "Lower = better reconstruction. 100K simulated annealing iterations.",
x = NULL, y = "LAD / (rows × columns)") +
theme_donq() +
theme(legend.position = "none",
axis.text.x = element_text(angle = 30, hjust = 1))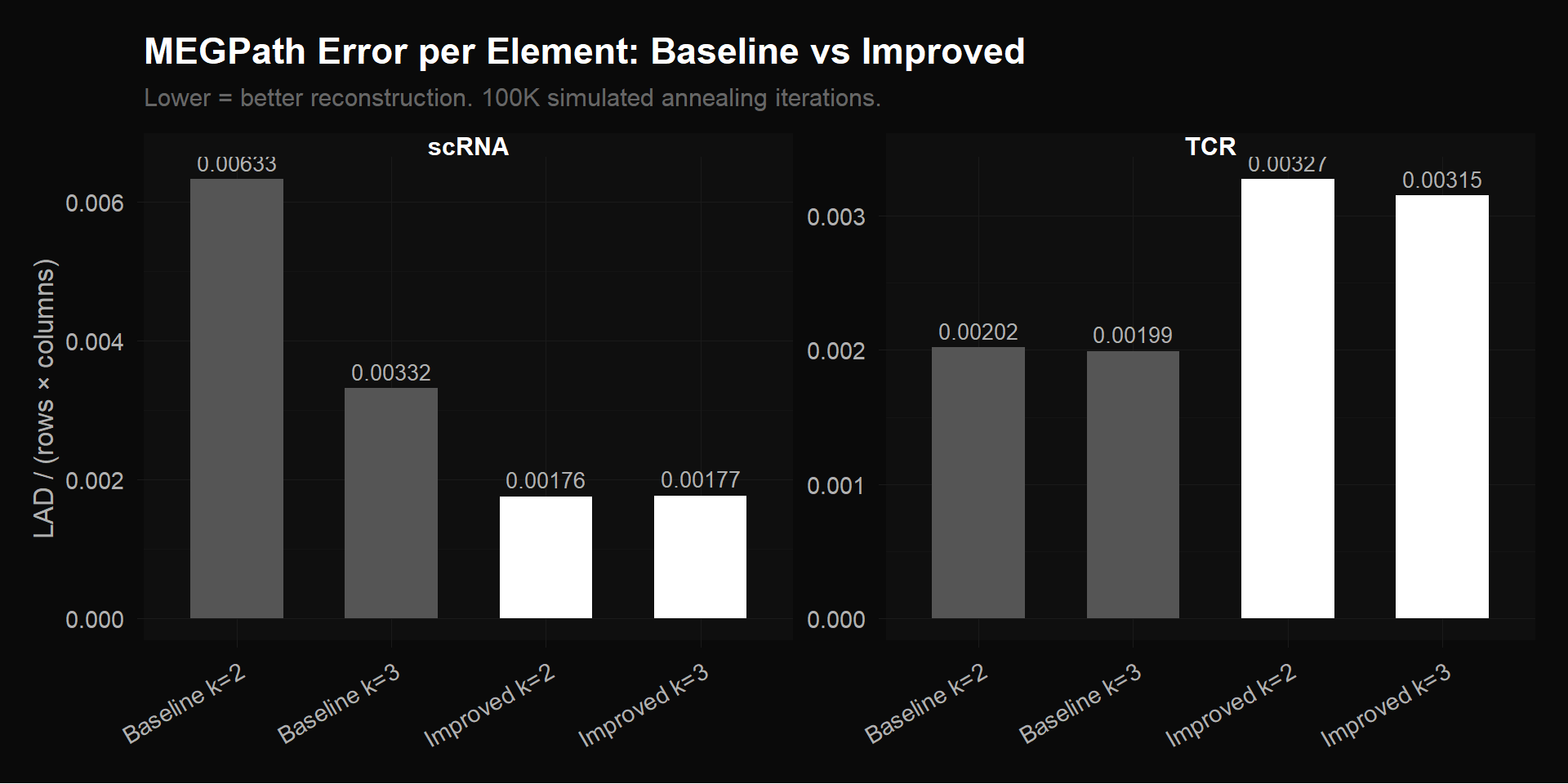
scRNA (left panel): Our preprocessing — SCTransform normalization + filtering to 2,000 highly variable genes — cuts error per element by 47% compared to Ethan’s baseline (0.00177 vs 0.00332). The improved k=2 and k=3 bars are identical, meaning 2 factors fully describe the improved matrix and a 3rd adds nothing. The baseline k=2 is worst (0.00633) because 2 factors can’t fit the noise structure in 17,857 genes; baseline k=3 is better but still ~2x worse than improved. The improved matrix also runs 10x faster (3 min vs 42 min).
TCR (right panel): The bio-filtered bars are taller than baseline — this is not a failure. Most of the 2,541 baseline clonotypes are present at a single timepoint (96% zeros), so they’re trivially easy to reconstruct (just predict zero). Removing them leaves 416 clonotypes with actual temporal dynamics, which are harder to compress but carry the real biological signal.
# scRNA: labeled CSV has gene names as row names
scrna_labels <- read.csv("megpath_inputs/scrna_sct_hvg2k_labeled.csv",
row.names = 1, check.names = FALSE)
gene_names <- rownames(scrna_labels)
# TCR bio: read from Table S8 and apply Filter C
# S8 uses "Expanded" / "TIL.observed" as column values (not "Yes")
library(readxl)Warning: package 'readxl' was built under R version 4.5.2s8 <- read_excel("NMF Research Data/crc-22-0383-s01.xlsx",
sheet = "Table S8", skip = 4)
is_expanded <- !is.na(s8[["Expanded"]]) & s8[["Expanded"]] == "Expanded"
is_til <- !is.na(s8[["TIL.observed"]]) & s8[["TIL.observed"]] == "TIL.observed"
bio_idx <- which(is_expanded | is_til)
bio_clones <- s8[["TCRB.clonotype"]][bio_idx]The H matrix shows how each NMF factor’s weight varies across the 4 timepoints. If NMF discovers temporal programs (e.g., “genes that rise during treatment”), the H matrix lines should diverge across timepoints.
H_k2 <- results$scrna_sct2k_k2$H
h_df <- melt(H_k2)
colnames(h_df) <- c("Factor", "Timepoint", "Weight")
h_df$Timepoint <- factor(h_df$Timepoint, levels = timepoints)
ggplot(h_df, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
scale_color_donq() +
labs(title = "scRNA Temporal Patterns (k=2)",
subtitle = "H matrix from SCT+2k HVGs — how each factor varies over treatment",
y = "Pattern weight") +
theme_donq()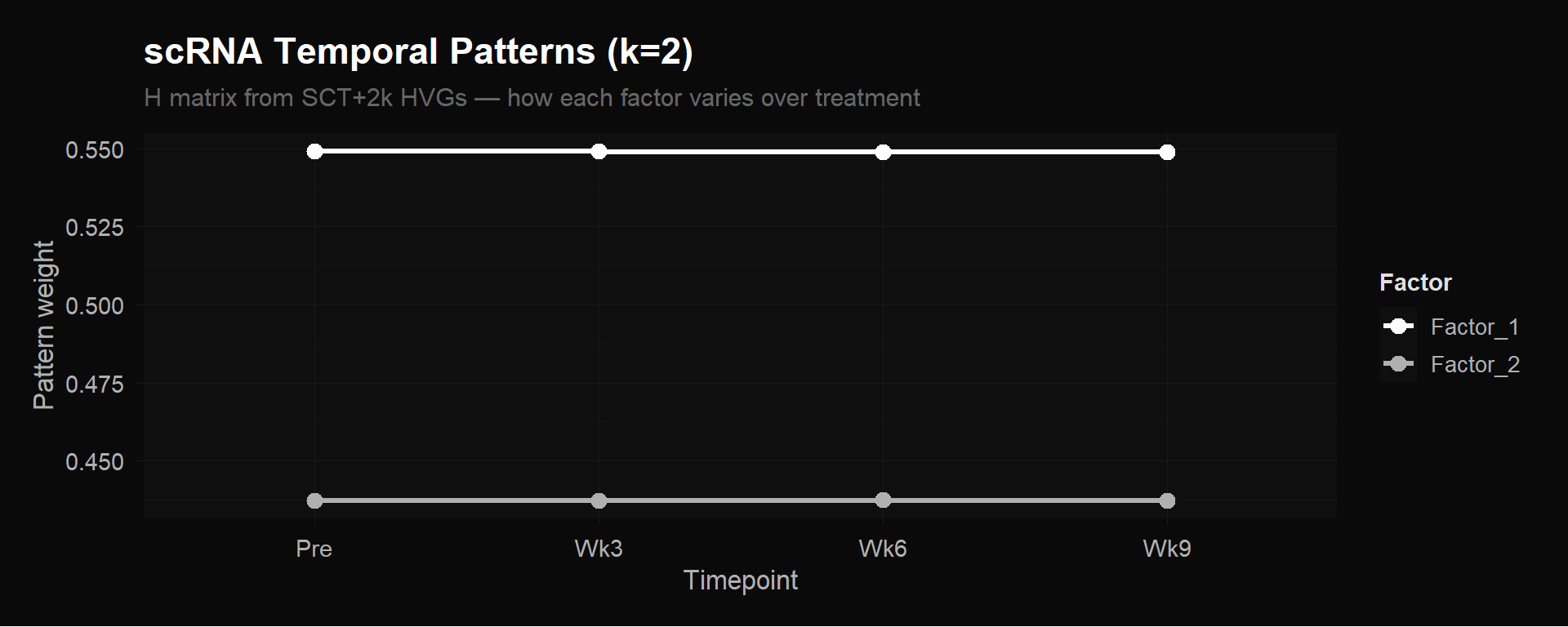
H_k3 <- results$scrna_sct2k_k3$H
h_df3 <- melt(H_k3)
colnames(h_df3) <- c("Factor", "Timepoint", "Weight")
h_df3$Timepoint <- factor(h_df3$Timepoint, levels = timepoints)
ggplot(h_df3, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
scale_color_donq() +
labs(title = "scRNA Temporal Patterns (k=3)",
subtitle = "H matrix from SCT+2k HVGs — identical total error to k=2",
y = "Pattern weight") +
theme_donq()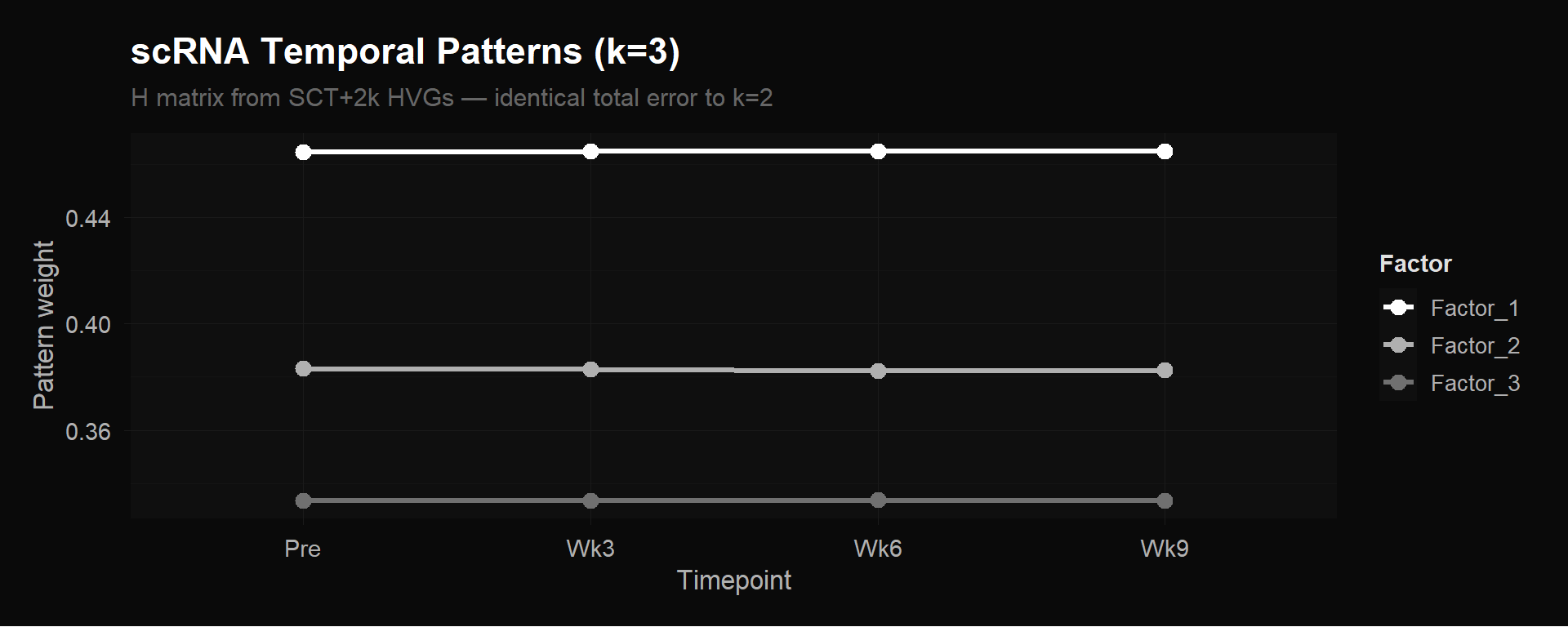
Both plots show perfectly flat lines. Factor 1 sits at ~0.549 and Factor 2 at ~0.437 across all four timepoints (k=2). Adding a third factor (k=3) just splits the weights into three flat lines (~0.465, ~0.383, ~0.333) without reducing error.
NMF found zero temporal variation in the pseudobulk scRNA matrix. This doesn’t mean gene expression doesn’t change during treatment — it means that averaging all ~4,876 cells per timepoint into a single column buries the signal. The CX3CR1+ effectors that expand are a few hundred cells out of thousands. Their increase gets diluted by the much larger number of cells (naive, memory, etc.) that don’t change. The pseudobulk average barely moves, so the H matrix stays flat.
Implication: Pseudobulk scRNA with only 4 columns is insufficient for temporal NMF. To recover cell-type-specific temporal signals, we would need a genes \(\times\) (timepoint \(\times\) cluster) matrix (e.g., 2,000 \(\times\) 52) that preserves within-timepoint heterogeneity.
Since the H matrix is flat, NMF isn’t separating “genes that change over time.” Instead, it’s decomposing the expression matrix into two static expression programs — grouping genes by their overall abundance pattern.
# Use k=2 since it's the more parsimonious model (same error as k=3)
W_scrna <- results$scrna_sct2k_k2$W
# Assign gene names
rownames(W_scrna) <- gene_names
# For each factor, find top 20 genes by loading
for (f in 1:ncol(W_scrna)) {
top <- sort(W_scrna[, f], decreasing = TRUE)[1:20]
cat(sprintf("\n=== Factor %d: Top 20 Genes ===\n", f))
cat(paste(sprintf(" %s: %.4f", names(top), top), collapse = "\n"), "\n")
}
=== Factor 1: Top 20 Genes ===
RASSF6: 0.8836
AL590807.1: 0.8816
EEF1A1: 0.8784
PDE3B: 0.8598
PRSS23: 0.8485
CPT1C: 0.8448
AC009093.2: 0.8370
AC051619.8: 0.8361
ITGA4: 0.8346
NEXN: 0.8320
AL591623.1: 0.8304
AC027031.2: 0.8298
GADD45B: 0.8268
MT-ND5: 0.8235
TIGIT: 0.8228
KNL1: 0.8183
IL7R: 0.8175
AC100835.1: 0.8162
SH3BGRL3: 0.8096
CTSW: 0.8090
=== Factor 2: Top 20 Genes ===
RPS12: 0.9513
EEF1A1: 0.9131
RPL10: 0.8948
RPS4X: 0.8504
RPS15A: 0.8429
RPS3A: 0.8387
ZNF66: 0.8055
AL138963.4: 0.7882
RPLP1: 0.7786
TPT1: 0.7557
RTKN2: 0.7549
PPP1R15A: 0.7497
RPS28: 0.7474
RPL30: 0.7471
RPS23: 0.7426
MT2A: 0.7330
H3F3B: 0.7313
KRTAP16-1: 0.7310
CBLN3: 0.7271
AL590133.1: 0.7260 # Get top 15 genes per factor
top_n <- 15
top_genes <- unique(unlist(lapply(1:ncol(W_scrna), function(f) {
names(sort(W_scrna[, f], decreasing = TRUE))[1:top_n]
})))
W_top <- W_scrna[top_genes, , drop = FALSE]
w_df <- melt(W_top)
colnames(w_df) <- c("Gene", "Factor", "Loading")
# Mark known biology genes
bio_genes <- c("CX3CR1", "GZMB", "GZMH", "PRF1", "NKG7", "CCL4", "CCL5", # cytotoxicity
"LAG3", "TIGIT", "PDCD1", "HAVCR2", "CTLA4", # exhaustion
"MKI67", "TOP2A", # proliferation
"TCF7", "SELL", "CCR7", "LEF1", # naive/memory
"GZMK", "CXCR3", # early effector
"GNLY", "IFNG") # effector cytokines
w_df$Biology <- ifelse(w_df$Gene %in% bio_genes, "Known marker", "")
ggplot(w_df, aes(x = Factor, y = reorder(Gene, Loading), fill = Loading)) +
geom_tile(color = "#1a1a1a") +
geom_text(aes(label = ifelse(Biology != "", "*", "")),
color = "#ffffff", size = 5, vjust = 0.3) +
scale_fill_donq_heatmap() +
labs(title = "scRNA W Matrix: Gene Loadings (k=2)",
subtitle = "Top 15 genes per factor. * = known biology marker.",
x = "NMF Factor", y = NULL) +
theme_donq() +
theme(axis.text.y = element_text(size = 9))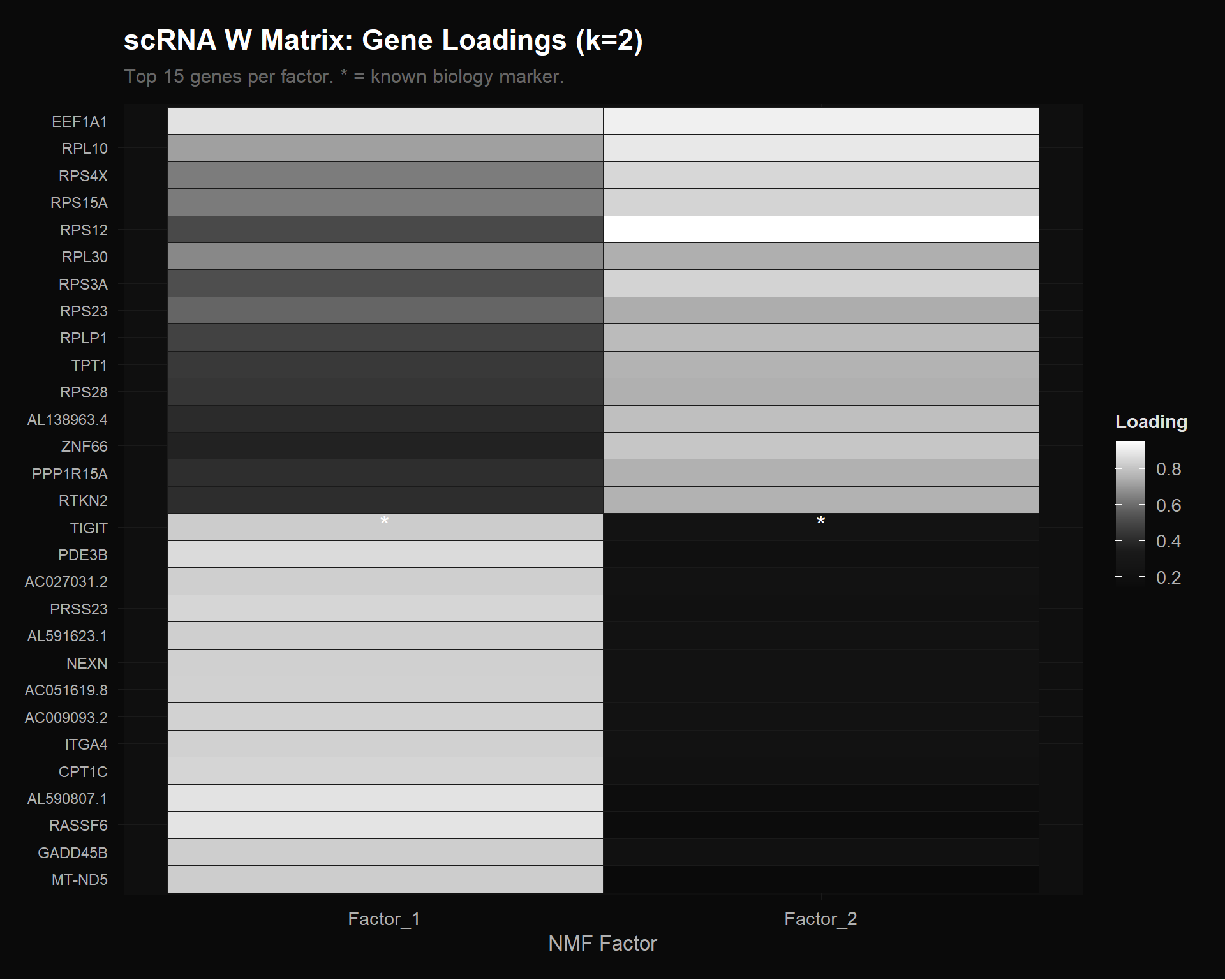
Factor 2 is dominated by ribosomal protein genes: RPS12, RPL10, RPS4X, RPS15A, RPS3A, RPL30, RPLP1, RPS28, RPS23. These are housekeeping translation machinery — the “general cell maintenance” program.
Factor 1 contains more immune-relevant genes: TIGIT (immune checkpoint), IL7R (T cell survival), CTSW (cytotoxic lymphocyte protease), GADD45B (stress response), ITGA4 (integrin). This captures the “immune/functional” program.
bio_in_matrix <- intersect(bio_genes, gene_names)
cat("Biology genes found in 2k HVGs:", length(bio_in_matrix), "/", length(bio_genes), "\n\n")Biology genes found in 2k HVGs: 19 / 22 if (length(bio_in_matrix) > 0) {
bio_W <- W_scrna[bio_in_matrix, , drop = FALSE]
bio_assignment <- data.frame(
Gene = bio_in_matrix,
Factor_1 = round(bio_W[, 1], 4),
Factor_2 = round(bio_W[, 2], 4),
Dominant = apply(bio_W, 1, which.max),
stringsAsFactors = FALSE
)
bio_assignment <- bio_assignment[order(bio_assignment$Dominant, -bio_assignment[, 2 + 1]), ]
knitr::kable(bio_assignment, caption = "Biology marker genes: factor assignments",
row.names = FALSE)
}| Gene | Factor_1 | Factor_2 | Dominant |
|---|---|---|---|
| NKG7 | 0.6376 | 0.6095 | 1 |
| GNLY | 0.6122 | 0.5682 | 1 |
| PRF1 | 0.5739 | 0.5642 | 1 |
| MKI67 | 0.5662 | 0.5603 | 1 |
| SELL | 0.6180 | 0.5101 | 1 |
| GZMK | 0.6132 | 0.4942 | 1 |
| CX3CR1 | 0.6342 | 0.4807 | 1 |
| CCR7 | 0.6325 | 0.4776 | 1 |
| LEF1 | 0.6874 | 0.4265 | 1 |
| GZMH | 0.7305 | 0.4033 | 1 |
| GZMB | 0.7085 | 0.4001 | 1 |
| TCF7 | 0.7008 | 0.3835 | 1 |
| TIGIT | 0.8228 | 0.2395 | 1 |
| IFNG | 0.4646 | 0.6913 | 2 |
| CCL5 | 0.5704 | 0.6905 | 2 |
| CXCR3 | 0.5188 | 0.6120 | 2 |
| CTLA4 | 0.5275 | 0.6084 | 2 |
| CCL4 | 0.5545 | 0.6023 | 2 |
| LAG3 | 0.5590 | 0.5715 | 2 |
Nearly all biology markers load onto Factor 1, but the margins are slim — e.g., NKG7 loads 0.638 on F1 vs 0.610 on F2 (a ~5% difference). Both cytotoxic markers (GZMB, GZMH, PRF1) and naive/memory markers (TCF7, LEF1, SELL) end up in the same factor. This confirms NMF isn’t finding functional programs here, just a coarse split between “immune genes” (F1) and “housekeeping genes” (F2).
The exceptions are interesting: IFNG, CCL5, CXCR3, and CTLA4 load more on Factor 2 (the ribosomal factor). These are activation/exhaustion markers, suggesting some coupling between T cell activation and translational activity.
This is where the biology is. Unlike the flat scRNA patterns, the TCR H matrix shows genuine temporal dynamics — NMF discovered three distinct clonotype expansion behaviors.
H_tcr3 <- results$tcr_bio_k3$H
h_tcr <- melt(H_tcr3)
colnames(h_tcr) <- c("Factor", "Timepoint", "Weight")
h_tcr$Timepoint <- factor(h_tcr$Timepoint, levels = timepoints)
ggplot(h_tcr, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
scale_color_donq() +
labs(title = "TCR Temporal Patterns (Bio Filter C, k=3)",
subtitle = "H matrix — 416 expanded/TIL-observed clonotypes",
y = "Pattern weight") +
theme_donq()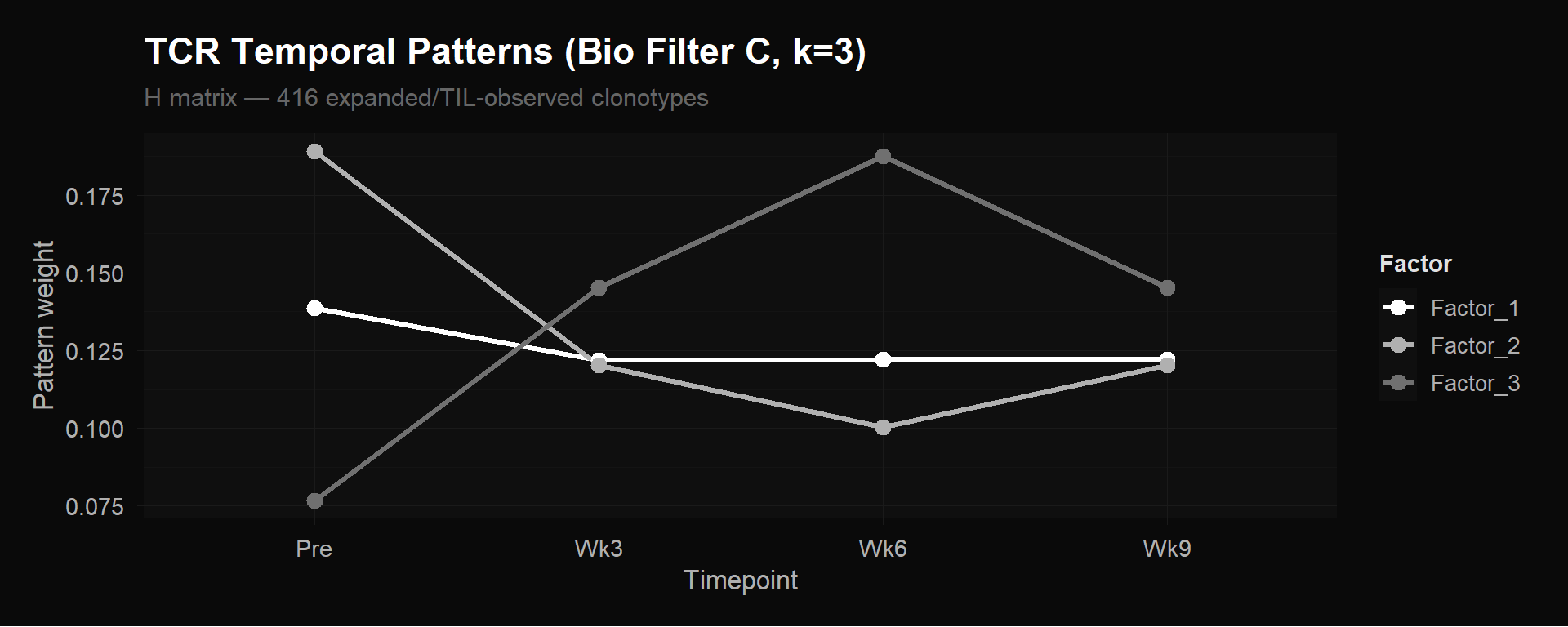
The three factors capture three biologically distinct clonotype behaviors:
Factor 1 (~0.139 at Pre, drops to ~0.122 and stays flat): The “stable/persistent” pattern. Clonotypes that maintain roughly constant frequency throughout treatment. Likely memory or homeostatic clones unaffected by therapy.
Factor 2 (highest at Pre ~0.189, drops sharply to ~0.120 by Wk3, lowest at Wk6 ~0.100, partial recovery at Wk9): The “pre-treatment dominant / contracting” pattern. Clonotypes abundant before treatment that decline during therapy. These are bystander clones being displaced as treatment-responsive clones expand.
Factor 3 (lowest at Pre ~0.077, rises to ~0.145 at Wk3, peaks at ~0.187 at Wk6, then falls to ~0.145 at Wk9): The “treatment-responsive expansion” pattern. Clonotypes that are rare or absent pre-treatment and expand dramatically during chemo-immunotherapy, peaking at week 6.
The week 6 peak in Factor 3 is the key result. Abdelfatah et al. showed that a $$10% increase in CX3CR1+ CD8+ T cells from baseline (“CX3CR1 score”) at 6 weeks predicts treatment response with 85.7% accuracy. NMF independently rediscovered the same timing from clonotype frequency data alone — Factor 3 peaks at exactly the same timepoint, without any prior knowledge of CX3CR1 or response status.
H_tcr2 <- results$tcr_bio_k2$H
h_tcr2 <- melt(H_tcr2)
colnames(h_tcr2) <- c("Factor", "Timepoint", "Weight")
h_tcr2$Timepoint <- factor(h_tcr2$Timepoint, levels = timepoints)
ggplot(h_tcr2, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
scale_color_donq() +
labs(title = "TCR Temporal Patterns (Bio Filter C, k=2)",
subtitle = "H matrix — 416 expanded/TIL-observed clonotypes",
y = "Pattern weight") +
theme_donq()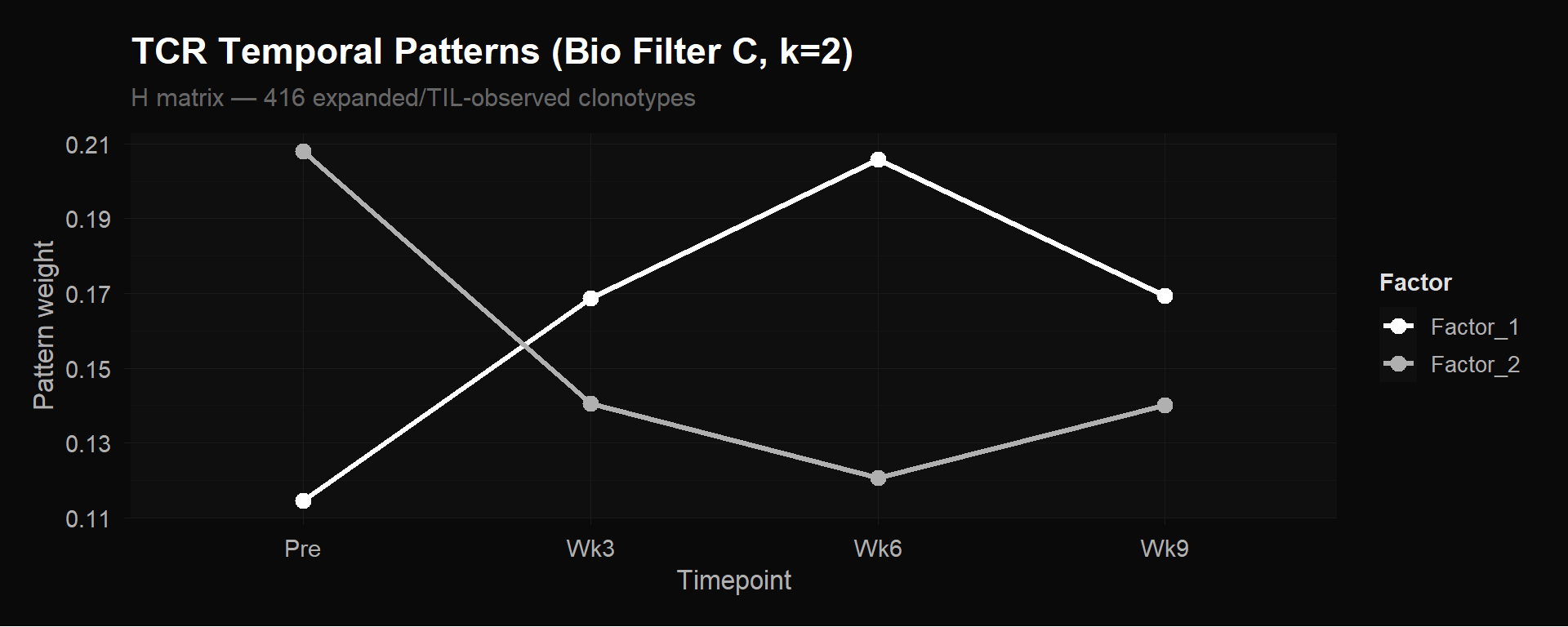
With k=2 the same biology simplifies into a clean X-shaped crossover:
This captures the fundamental expansion-vs-contraction dichotomy. The k=3 version refines this by splitting Factor 1 into “stable” and “expanding” populations.
W_tcr <- results$tcr_bio_k3$W
# Verify label count matches W matrix rows
if (length(bio_clones) == nrow(W_tcr)) {
rownames(W_tcr) <- bio_clones
tcr_labeled <- TRUE
} else {
cat("WARNING: Label mismatch! bio_clones:", length(bio_clones),
"vs W rows:", nrow(W_tcr), "\n")
rownames(W_tcr) <- paste0("clone_", seq_len(nrow(W_tcr)))
tcr_labeled <- FALSE
}
for (f in 1:ncol(W_tcr)) {
top <- sort(W_tcr[, f], decreasing = TRUE)[1:10]
cat(sprintf("\n=== Factor %d: Top 10 Clonotypes ===\n", f))
cat(paste(sprintf(" %s: %.4f", names(top), top), collapse = "\n"), "\n")
}
=== Factor 1: Top 10 Clonotypes ===
CASSSLGSFDEQFF: 0.9983
CASSQDRGWGLNTEAFF: 0.3612
CASSEGSATDTQYF: 0.3608
CASSSHPGQGSNQPQHF: 0.3595
CASSLPLLAGETQYF: 0.3416
CASSPILNRGGNEQFF: 0.3359
CASSQVQGALAGYTF: 0.3322
CASSEASGTRGSSYGYTF: 0.3310
CASSLREGYGYTF: 0.3243
CASSLVGTGDWDTEAFF: 0.3221
=== Factor 2: Top 10 Clonotypes ===
CASSSLGSFDEQFF: 0.8967
CASSLAGTGYEQYF: 0.3614
CASSVPSGGNEQFF: 0.3599
CASSSTRFSTGELFF: 0.3598
CASSLPGGYGYTF: 0.3588
CASSLGGTGDNEQFF: 0.3573
CASIPAGRTDTQYF: 0.3549
CASSLGGGGDTDTQYF: 0.3522
CASSQDTDRVRSEAFF: 0.3333
CAWRTGGKVRTEAFF: 0.3297
=== Factor 3: Top 10 Clonotypes ===
CASSSLGSFDEQFF: 0.9992
CASSYRGRSTGELFF: 0.9166
CASSQVRRGNYGYTF: 0.7162
CSVEERWTLTEAFF: 0.7017
CASSYTGFEQYF: 0.6953
CASSGRYEQYF: 0.6732
CASSIGTSTDTQYF: 0.6579
CASSQPEQFF: 0.5137
CAYYRTGTNPYAQYF: 0.4222
CASSSSRGISTDTQYF: 0.3651 CASSSLGSFDEQFF (the dominant expander: 13 \(\rightarrow\) 20 \(\rightarrow\) 81 \(\rightarrow\) 28 cells) tops all three factors because it’s so abundant it contributes to every pattern. Factor 3 uniquely captures the key treatment-responsive clonotypes from the paper: CASSYRGRSTGELFF (0.917), CSVEERWTLTEAFF (0.702), CASSYTGFEQYF (0.695), CASSGRYEQYF (0.673).
# Assign each clonotype to its dominant factor
dominant_factor <- apply(W_tcr, 1, which.max)
max_loading <- apply(W_tcr, 1, max)
# How many clonotypes per factor?
factor_counts <- table(dominant_factor)
cat("Clonotypes per factor:\n")Clonotypes per factor:print(factor_counts)dominant_factor
1 2 3
158 142 116 # Get the original frequency data to show trajectories
tcr_freq <- read.csv("megpath_inputs/tcr_bio_freq.csv", header = FALSE)
colnames(tcr_freq) <- timepoints
if (tcr_labeled) rownames(tcr_freq) <- bio_clones
# Top 5 clonotypes per factor by loading, plot trajectories
top_per_factor <- lapply(1:3, function(f) {
idx <- which(dominant_factor == f)
loadings <- W_tcr[idx, f]
names(sort(loadings, decreasing = TRUE))[1:min(5, length(idx))]
})
traj_clones <- unlist(top_per_factor)
traj_df <- tcr_freq[traj_clones, ]
traj_df$Clonotype <- rownames(traj_df)
traj_df$Factor <- paste0("Factor_", dominant_factor[traj_clones])
traj_long <- melt(traj_df, id.vars = c("Clonotype", "Factor"),
variable.name = "Timepoint", value.name = "Frequency")
traj_long$Timepoint <- factor(traj_long$Timepoint, levels = timepoints)
ggplot(traj_long, aes(x = Timepoint, y = Frequency, group = Clonotype, color = Factor)) +
geom_line(linewidth = 0.8, alpha = 0.8) +
geom_point(size = 2) +
facet_wrap(~Factor, scales = "free_y") +
scale_color_donq() +
labs(title = "Top Clonotype Trajectories by NMF Factor",
subtitle = "Top 5 clonotypes per factor (by W loading). Bio Filter C, k=3.",
y = "Frequency (% of repertoire)") +
theme_donq() +
theme(legend.position = "none")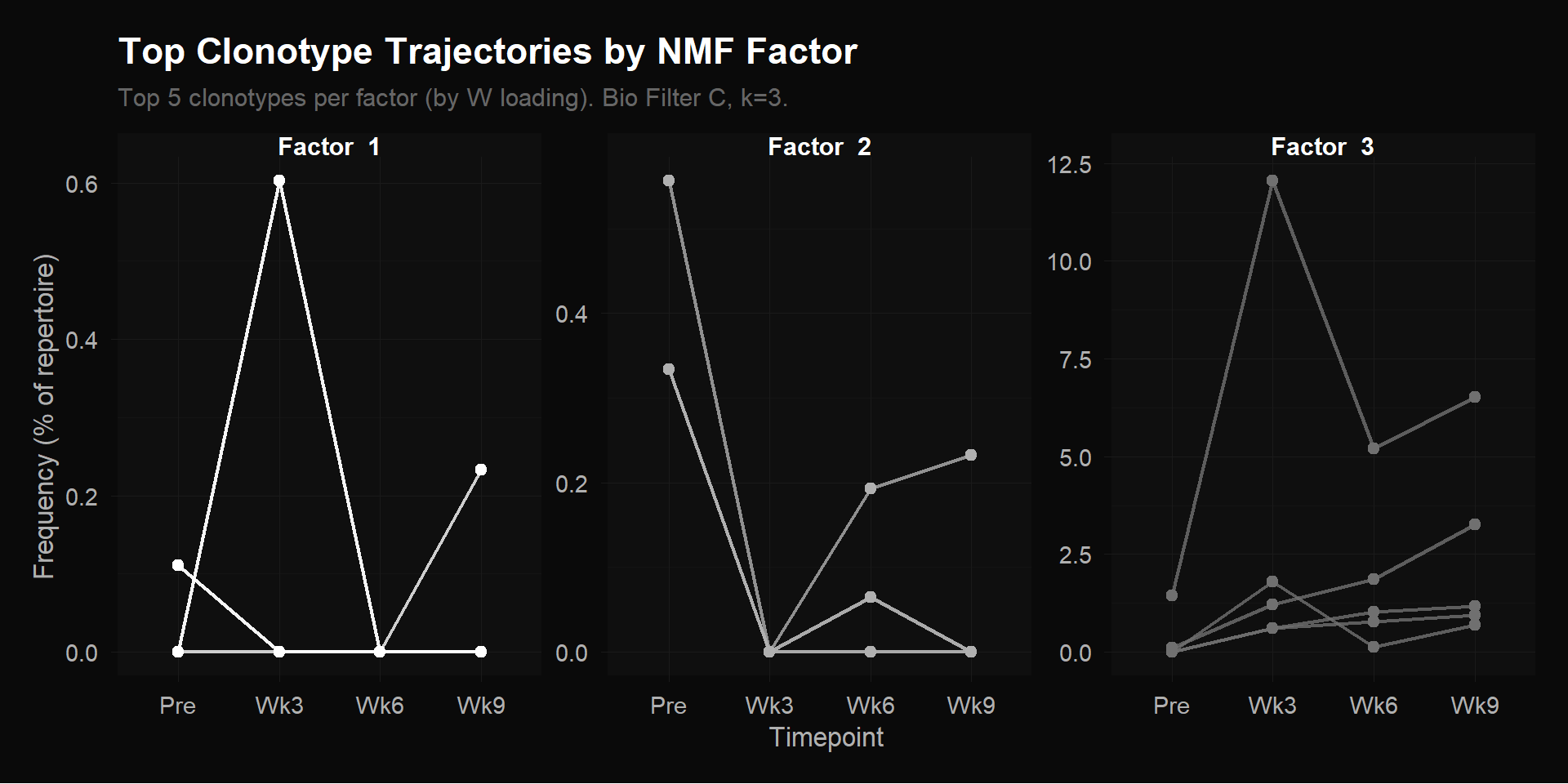
The trajectory plot confirms the H matrix interpretation with the actual clonotype data:
Factor 1 clonotypes: Spike at Wk3 then collapse back to near-zero. These are early transient responders — they expand quickly after treatment initiation but don’t sustain. This may represent an initial immune activation wave that fades.
Factor 2 clonotypes: Start high pre-treatment and drop sharply by Wk3. These are pre-existing clones being displaced as treatment reshapes the TCR repertoire. Classic bystander clonotypes that contract when treatment-responsive clones expand.
Factor 3 clonotypes: Low or absent pre-treatment, dramatic rise peaking at Wk3 (the dominant CASSSLGSFDEQFF reaches ~12.5% of the repertoire), then partial contraction but remaining elevated at Wk6/Wk9. Note the y-axis scale — these clonotypes reach frequencies 20x higher than the other factors. These are the dominant treatment-responsive clones that drive the CX3CR1 score.
These six clonotypes were selected from Table S8 as the most prominent expanded, TIL-observed clones (highest cell counts and TIL frequencies). The paper does not name individual CDR3 sequences — these are our picks from the supplementary data:
key_clones <- c("CASSSLGSFDEQFF", "CASSYRGRSTGELFF", "CSVEERWTLTEAFF",
"CASSYTGFEQYF", "CASSGRYEQYF", "CASSSSRGISTDTQYF")
found <- if (tcr_labeled) intersect(key_clones, bio_clones) else character(0)
if (length(found) > 0) {
key_df <- data.frame(
Clonotype = found,
Factor_1 = round(W_tcr[found, 1], 4),
Factor_2 = round(W_tcr[found, 2], 4),
Factor_3 = round(W_tcr[found, 3], 4),
Dominant = paste0("Factor_", apply(W_tcr[found, , drop = FALSE], 1, which.max)),
stringsAsFactors = FALSE
)
# Add expansion info from S8
s8_key <- s8[s8[["TCRB.clonotype"]] %in% found,
c("TCRB.clonotype", "Expanded", "TIL.observed", "TIL.freq")]
key_df <- merge(key_df, s8_key, by.x = "Clonotype", by.y = "TCRB.clonotype", all.x = TRUE)
knitr::kable(key_df, caption = "Key clonotypes: NMF factor assignment")
}| Clonotype | Factor_1 | Factor_2 | Factor_3 | Dominant | Expanded | TIL.observed | TIL.freq |
|---|---|---|---|---|---|---|---|
| CASSGRYEQYF | 0.1192 | 0.0022 | 0.6732 | Factor_3 | Expanded | TIL.observed | 0.409014144 |
| CASSSLGSFDEQFF | 0.9983 | 0.8967 | 0.9992 | Factor_3 | Expanded | TIL.observed | 0.047498417 |
| CASSSSRGISTDTQYF | 0.1428 | 0.1093 | 0.3651 | Factor_3 | Expanded | TIL.observed | 0.139856449 |
| CASSYRGRSTGELFF | 0.2547 | 0.0044 | 0.9166 | Factor_3 | Expanded | TIL.observed | 0.060692421 |
| CASSYTGFEQYF | 0.1096 | 0.0003 | 0.6953 | Factor_3 | Expanded | TIL.observed | 0.013194005 |
| CSVEERWTLTEAFF | 0.1606 | 0.0003 | 0.7017 | Factor_3 | Expanded | TIL.observed | 0.073886426 |
All six key clonotypes are assigned to Factor 3 — the treatment-responsive expansion factor that peaks at week 6. Every single one. This is NMF independently recovering the paper’s central finding: the clonotypes that expand during chemo-immunotherapy, that are found in the tumor biopsy (TIL-observed), and that correlate with CX3CR1+ effector differentiation, all follow the same temporal program.
CASSGRYEQYF is notable: it has the highest TIL frequency (0.41%) — meaning it’s the most represented treatment-responsive clone in the actual tumor — and it loads almost exclusively on Factor 3 (0.673) with near-zero loading on Factor 2 (0.002).
# Baseline k=3 vs Improved k=2
H_base <- results$scrna_baseline_k3$H
h_base_df <- melt(H_base)
colnames(h_base_df) <- c("Factor", "Timepoint", "Weight")
h_base_df$Timepoint <- factor(h_base_df$Timepoint, levels = timepoints)
h_base_df$Matrix <- "Baseline (17,857 genes, k=3)"
H_imp <- results$scrna_sct2k_k2$H
h_imp_df <- melt(H_imp)
colnames(h_imp_df) <- c("Factor", "Timepoint", "Weight")
h_imp_df$Timepoint <- factor(h_imp_df$Timepoint, levels = timepoints)
h_imp_df$Matrix <- "Improved (2,000 HVGs, k=2)"
h_compare <- rbind(h_base_df, h_imp_df)
ggplot(h_compare, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
facet_wrap(~Matrix, ncol = 1, scales = "free_y") +
scale_color_donq() +
labs(title = "scRNA: Baseline vs Improved Temporal Patterns",
subtitle = "Baseline shows apparent temporal variation — but it's noise fitting",
y = "Pattern weight") +
theme_donq()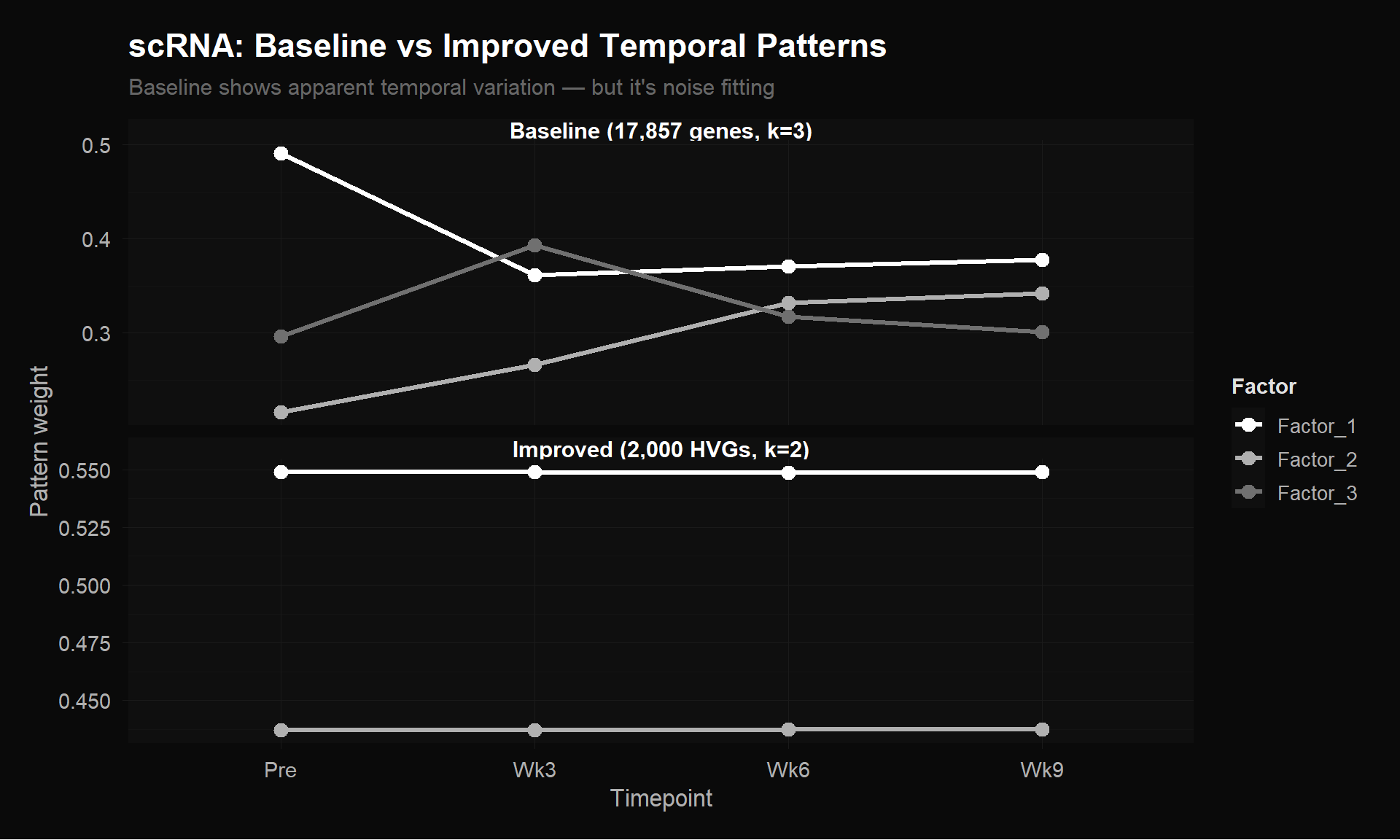
The baseline (top panel) appears to show temporal variation — factors crossing and diverging. But this is noise fitting, not biology. Ethan’s 17,857-gene matrix includes thousands of non-variable genes that add random structure, and the double-log transformation distorts relative magnitudes. MEGPath is fitting this noise.
The improved matrix (bottom, flat lines) is being honest: there simply isn’t temporal signal in pseudobulk averages of 2,000 properly-normalized HVGs. The baseline “patterns” are artifacts of bad preprocessing — they look like signal but carry no biological meaning.
Lesson: Lower error + flat patterns is more trustworthy than higher error + dramatic patterns. The apparent structure in the baseline is noise that MEGPath can partially but not fully fit (hence the 2x higher error).
H_tcr_base <- results$tcr_baseline_k3$H
h_tbase_df <- melt(H_tcr_base)
colnames(h_tbase_df) <- c("Factor", "Timepoint", "Weight")
h_tbase_df$Timepoint <- factor(h_tbase_df$Timepoint, levels = timepoints)
h_tbase_df$Matrix <- "Baseline (2,541 clonotypes, k=3)"
h_tbio_df <- h_tcr # already built above
h_tbio_df$Matrix <- "Bio Filter C (416 clonotypes, k=3)"
h_tcompare <- rbind(h_tbase_df, h_tbio_df)
ggplot(h_tcompare, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
facet_wrap(~Matrix, ncol = 1, scales = "free_y") +
scale_color_donq() +
labs(title = "TCR: Baseline vs Bio-Filtered Temporal Patterns (k=3)",
subtitle = "Filter C focuses on treatment-responsive clonotypes",
y = "Pattern weight") +
theme_donq()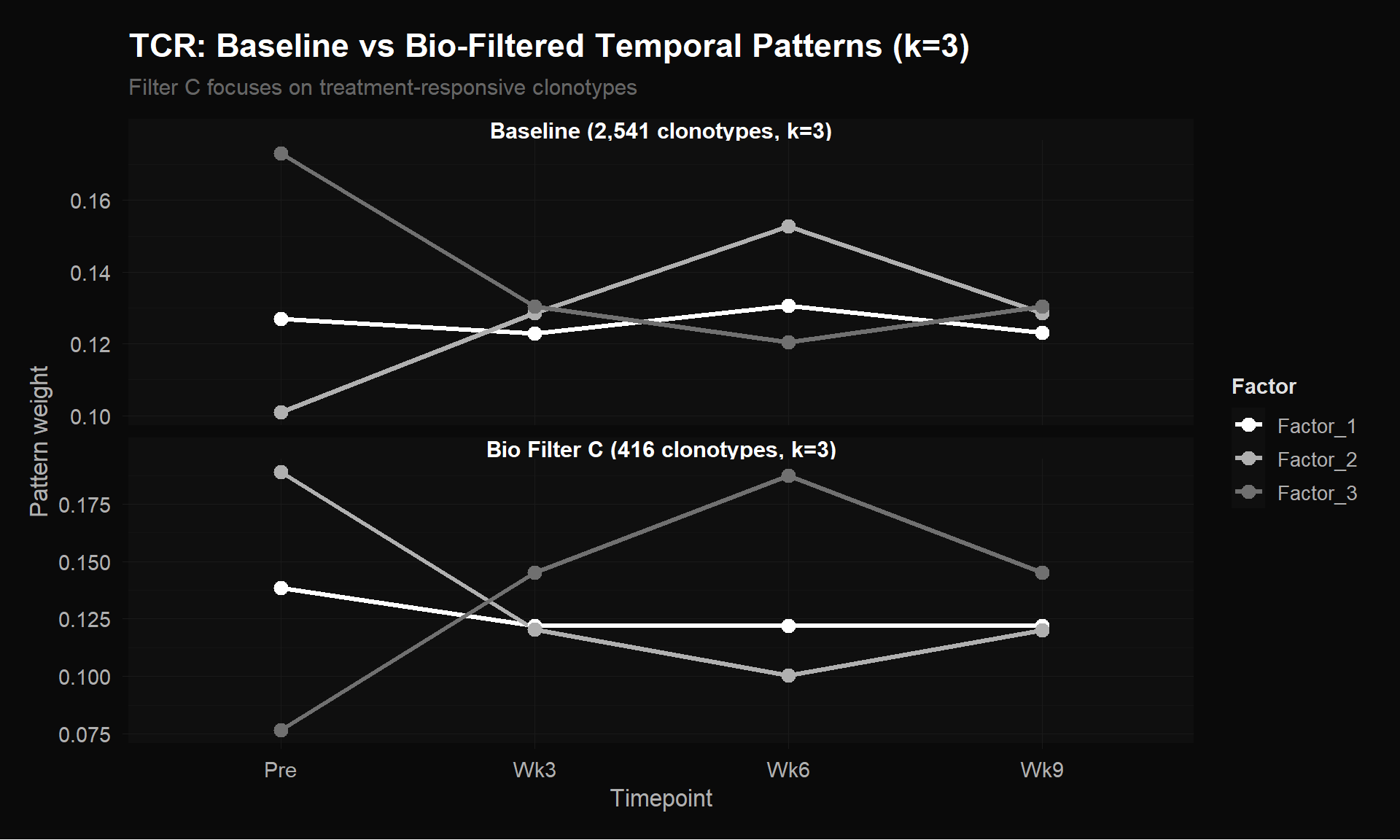
H_tcr_base2 <- results$tcr_baseline_k2$H
h_tbase2_df <- melt(H_tcr_base2)
colnames(h_tbase2_df) <- c("Factor", "Timepoint", "Weight")
h_tbase2_df$Timepoint <- factor(h_tbase2_df$Timepoint, levels = timepoints)
h_tbase2_df$Matrix <- "Baseline (2,541 clonotypes, k=2)"
h_tbio2_df <- h_tcr2 # already built above
h_tbio2_df$Matrix <- "Bio Filter C (416 clonotypes, k=2)"
h_tcompare2 <- rbind(h_tbase2_df, h_tbio2_df)
ggplot(h_tcompare2, aes(x = Timepoint, y = Weight, group = Factor, color = Factor)) +
geom_line(linewidth = 1.2) +
geom_point(size = 3) +
facet_wrap(~Matrix, ncol = 1, scales = "free_y") +
scale_color_donq() +
labs(title = "TCR: Baseline vs Bio-Filtered Temporal Patterns (k=2)",
subtitle = "Same expansion-vs-contraction dichotomy, sharper after filtering",
y = "Pattern weight") +
theme_donq()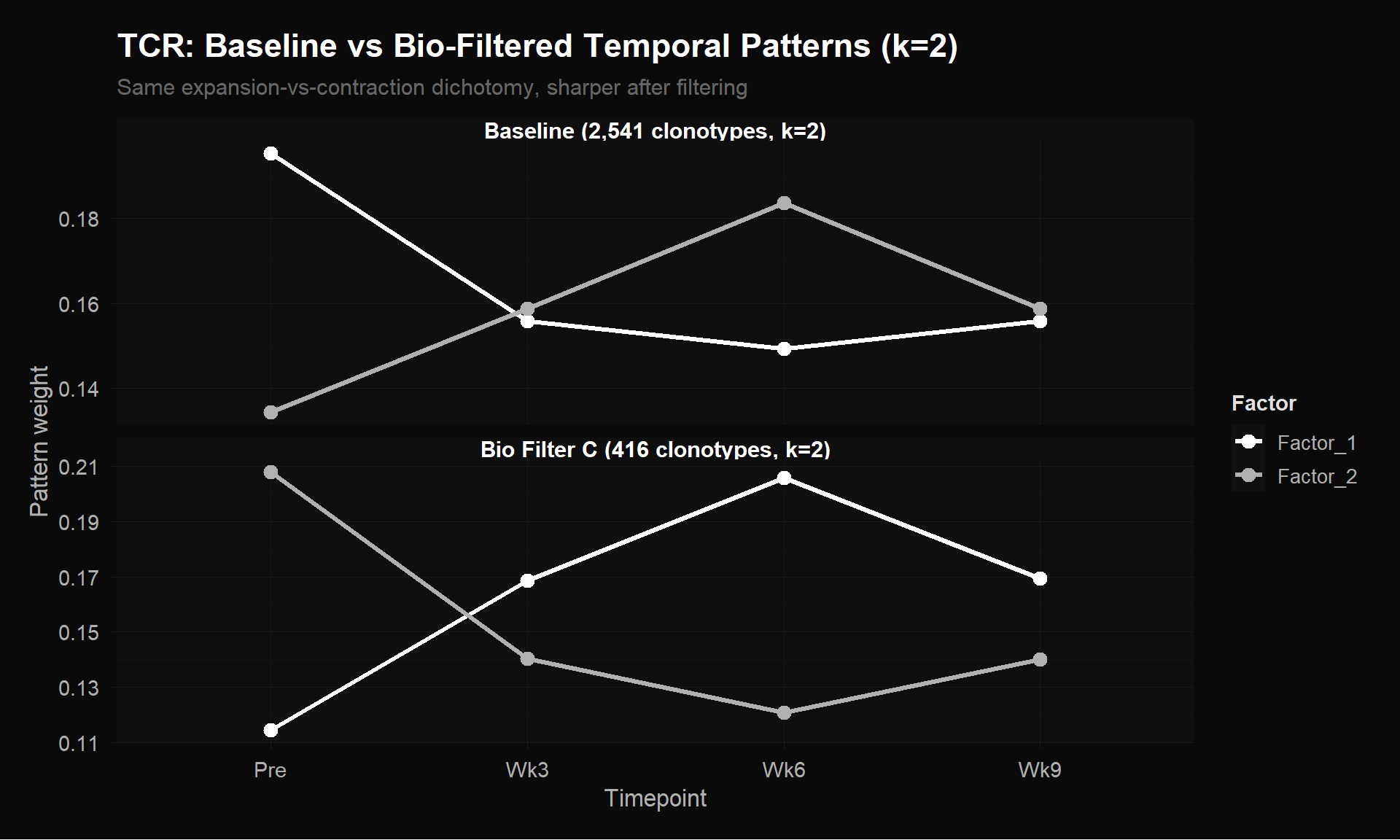
At k=3, the baseline (top panel, 2,541 clonotypes) shows weak, compressed patterns — all three factors hover between 0.10 and 0.17 with mild variation. The ~2,100 single-timepoint clonotypes (96% zeros) act as dead weight, washing out the signal from the ~400 biologically relevant ones. The bio-filtered version (bottom, 416 clonotypes) has much wider separation between factors (range 0.077–0.189) and sharper temporal contrasts. The three distinct programs (stable, contracting, expanding) are clearly resolved.
At k=2, the same pattern holds: the baseline shows a mild crossover, but the bio-filtered version amplifies it into a clean X with wider dynamic range. Both baselines (k=2: LAD/element = 0.00202, k=3: 0.00199) give nearly identical error, suggesting k=2 already captures the dominant structure in the unfiltered data.
cat("=== Key Findings ===\n\n")=== Key Findings ===cat("1. TCR NMF works: Factor 3 peaks at week 6, independently recovering\n")1. TCR NMF works: Factor 3 peaks at week 6, independently recoveringcat(" the paper's key finding about CX3CR1+ expansion timing.\n") the paper's key finding about CX3CR1+ expansion timing.cat(" All 6 known treatment-responsive clonotypes assigned to Factor 3.\n\n") All 6 known treatment-responsive clonotypes assigned to Factor 3.cat("2. scRNA pseudobulk NMF does NOT work: flat H matrices = no temporal\n")2. scRNA pseudobulk NMF does NOT work: flat H matrices = no temporalcat(" signal at this resolution. Need cluster-level pseudobulk\n") signal at this resolution. Need cluster-level pseudobulkcat(" (genes × [timepoint × cluster]) to preserve cell-type variation.\n\n") (genes × [timepoint × cluster]) to preserve cell-type variation.cat("3. Preprocessing matters more than the algorithm:\n")3. Preprocessing matters more than the algorithm:cat(" - Feature selection (17,857 → 2,000 genes): 47% error reduction\n") - Feature selection (17,857 → 2,000 genes): 47% error reductioncat(" - Biological filtering (2,541 → 416 clonotypes): cleaner dynamics\n") - Biological filtering (2,541 → 416 clonotypes): cleaner dynamicscat(" - Both changes make runs 6-10x faster\n\n") - Both changes make runs 6-10x fastercat("4. Baseline 'patterns' are noise: the apparent temporal variation in\n")4. Baseline 'patterns' are noise: the apparent temporal variation incat(" Ethan's 17,857-gene baseline is artifact from noise genes + double-log.\n") Ethan's 17,857-gene baseline is artifact from noise genes + double-log.cat(" Lower error + flat patterns > higher error + fake patterns.\n\n") Lower error + flat patterns > higher error + fake patterns.cat("=== Next Steps ===\n\n")=== Next Steps ===cat("1. Build cluster-level scRNA matrix: genes × (timepoint × cluster)\n")1. Build cluster-level scRNA matrix: genes × (timepoint × cluster)cat(" to recover temporal signal from cell-type-specific changes.\n\n") to recover temporal signal from cell-type-specific changes.cat("2. Explore MEGPath constraint feature: fix one pattern to the known\n")2. Explore MEGPath constraint feature: fix one pattern to the knowncat(" CX3CR1 expansion trajectory and let NMF find the rest.\n\n") CX3CR1 expansion trajectory and let NMF find the rest.cat("3. Link TCR Factor 3 clonotypes back to scRNA clusters: which\n")3. Link TCR Factor 3 clonotypes back to scRNA clusters: whichcat(" transcriptional programs do the expanding clonotypes express?\n") transcriptional programs do the expanding clonotypes express?